Label-free quantification of metabolites¶
Introduction¶
Quantification and identification of chemical compounds are basic tasks in metabolomic studies. In this tutorial session we construct a UPLC-MS-based, label-free quantification and identification workflow. Following quantification and identification we then perform statistical downstream analysis to detect quantification values that differ significantly between two conditions. This approach can, for example, be used to detect biomarkers.
Here, we analyze a dataset derived from bacterial cytosolic fractions to investigate the metabolic effects of fosfomycin, an antibiotic that inhibits a key step in peptidoglycan biosynthesis. The study is based on Bacillus subtilis cultures subjected to different treatment conditions.
The dataset consists of four sample types:
Blank – A sample without any analyte
Control – An untreated B. subtilis sample, representing baseline metabolic activity.
Treatment – A B. subtilis sample treated with fosfomycin
Pool – A composite sample combining aliquots from the three other conditions
The primary goal of this analysis is to evaluate the metabolic response of B. subtilis to fosfomycin treatment, with a specific focus on its effects on peptidoglycan biosynthesis pathways.
Basics of non-targeted metabolomics data analysis¶
For the metabolite quantification we choose an approach similar to the one used for peptides, but this time based on the OpenMS FeatureFinderMetabo method. This feature finder again collects peak-picked data into individual mass traces. The reason
why we need a different feature finder for metabolites lies in the step after trace detection: the aggregation of isotopic traces belonging to the same compound ion into the same feature. Compared to peptides with their averagine model, small molecules
have very different isotopic distributions. To group small molecule mass traces correctly, an aggregation model tailored to small molecules is thus needed.
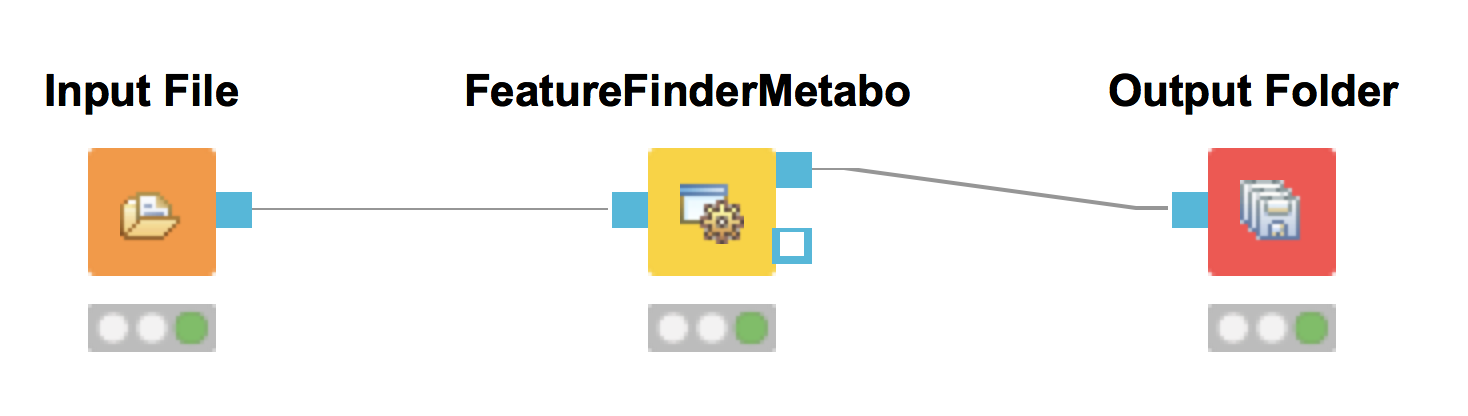
Create a new workflow called for instance ”Metabolomics”.
Add an
File Importernode and configure it with one mzML file from theExample_Data▸Metabolomics▸datasets .Add a
FeatureFinderMetabonode (from Community Nodes > OpenMS > Quantitation) and connect the first output port of theFile Importerto theFeatureFinderMetabo.For an optimal result adjust the following settings. Please note that some of these are advanced parameters.
Connect a
Output Folderto the output of theFeatureFinderMetabo(see Fig. 27).
|
|---|
Figure 27: FeatureFinderMetabo workflow |
In the following advanced parameters will be highlighted. These parameter can be altered if the Show advanced parameter field in the specific tool is activated.
parameter |
value |
|---|---|
algorithm→common→noise_threshold_int |
1000.0 |
algorithm→common→chrom_peak_snr |
3.0 |
algorithm→common→chrom_fwhm |
5.0 |
algorithm→mtd→mass_error_ppm |
10.0 |
algorithm→ffm→isotope_filtering_model |
‘none’ |
algorithm→ffm→remove_single_traces |
true |
algorithm→ffm→report_convex_hulls |
true |
The parameters change the behavior of FeatureFinderMetabo as follows:
noise_threshold_int: Intensity threshold below which peaks are regarded as noise.
chrom_peak_snr: Minimum signal-to-noise a mass trace should have.
chrom_fwhm: The expected chromatographic peak width in seconds.
mass_error_ppm: Allowed mass deviation (in ppm)
isotope_filtering_model: Remove/score candidate assemblies based on isotope intensities. SVM isotope models for metabolites were trained with either 2% or 5% RMS error. For peptides, an averagine cosine scoring is used. Select the appropriate noise model according to the quality of measurement or MS device. (valid: ‘metabolites (2% RMS)’, ‘metabolites (5% RMS)’, ‘peptides’, ‘none’)
remove_single_traces: If set to true, unassembled traces are removed (single traces).
report_convex_hulls: If set to true, convex hulls including mass traces will be reported for all identified features. This increases the output size considerably.
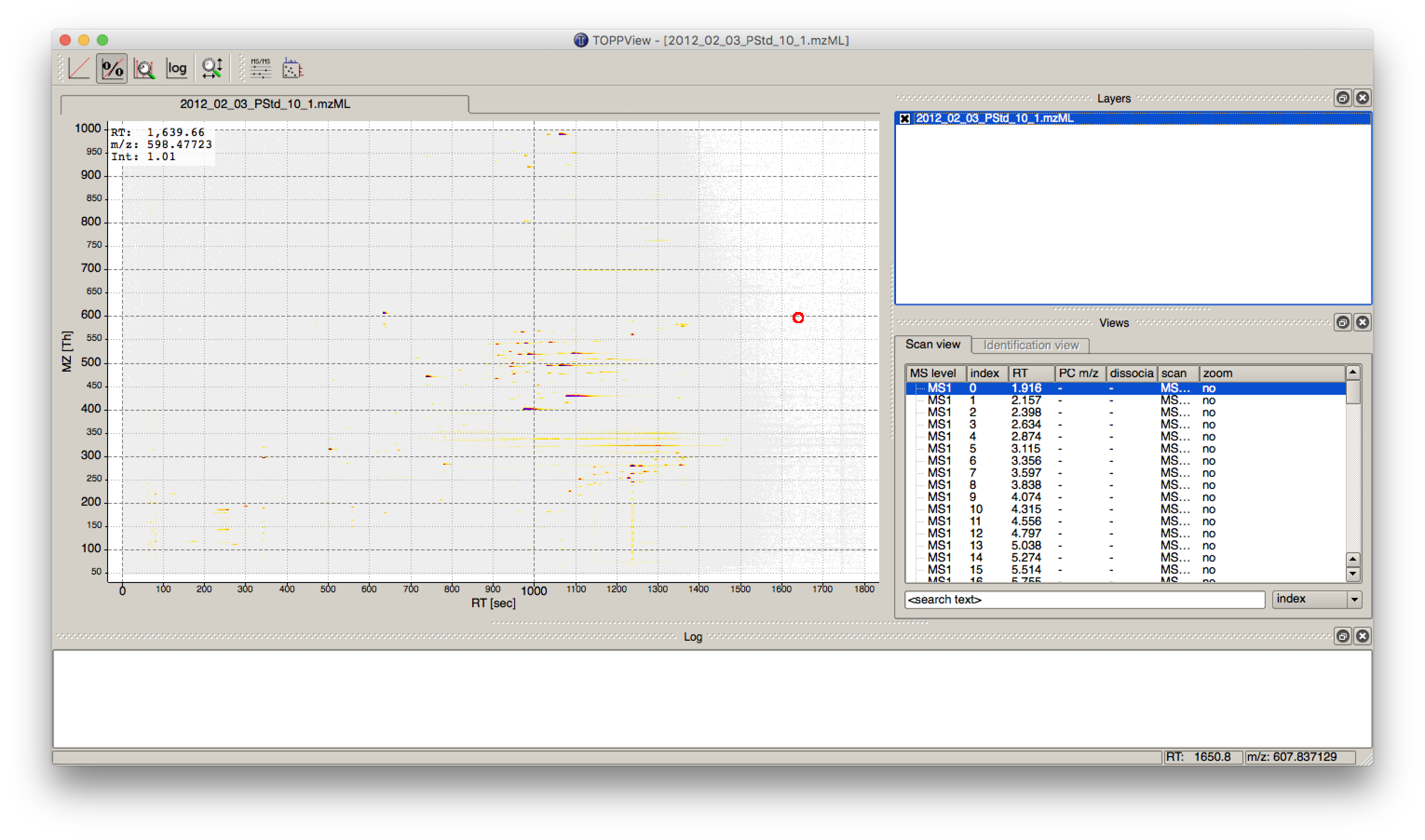
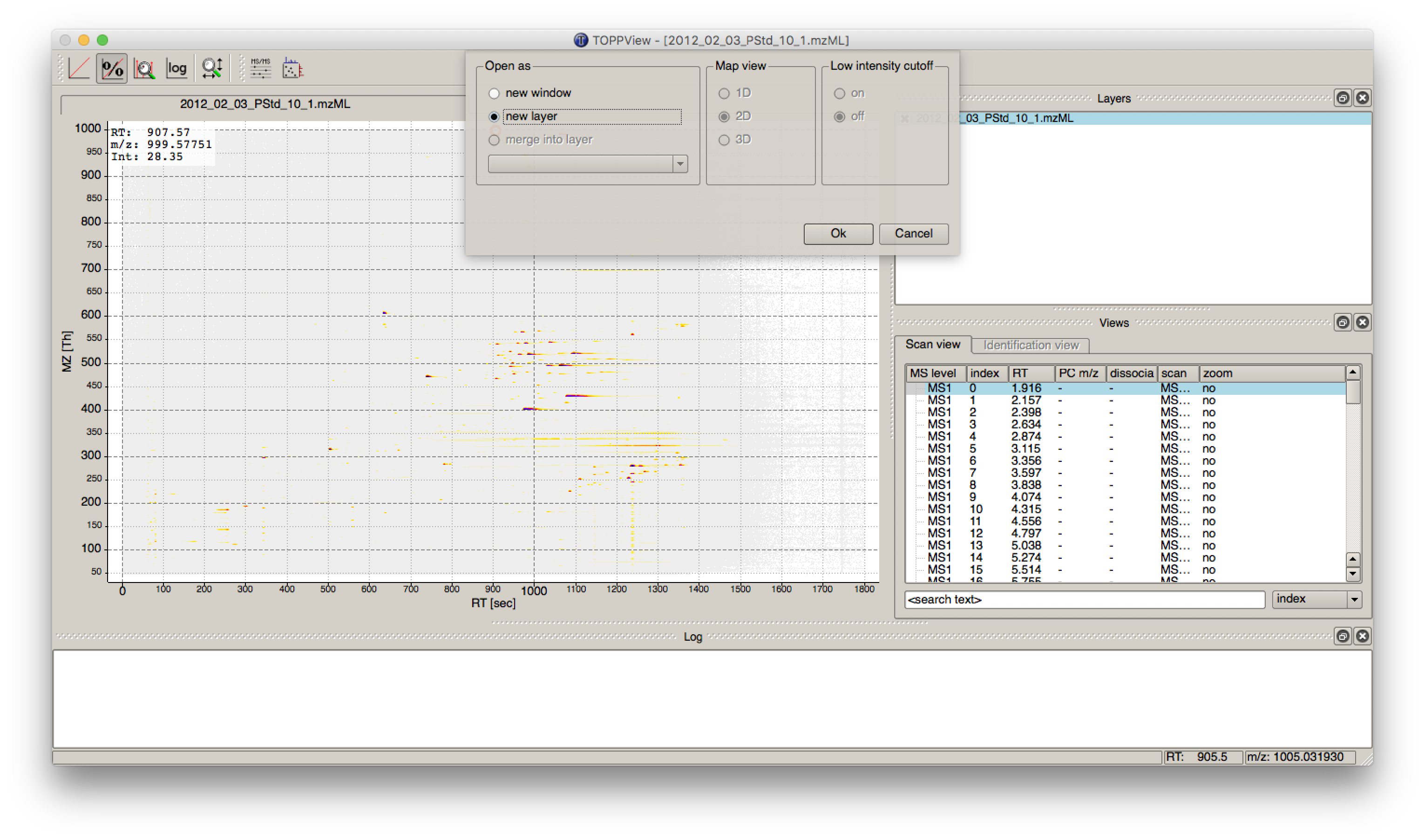
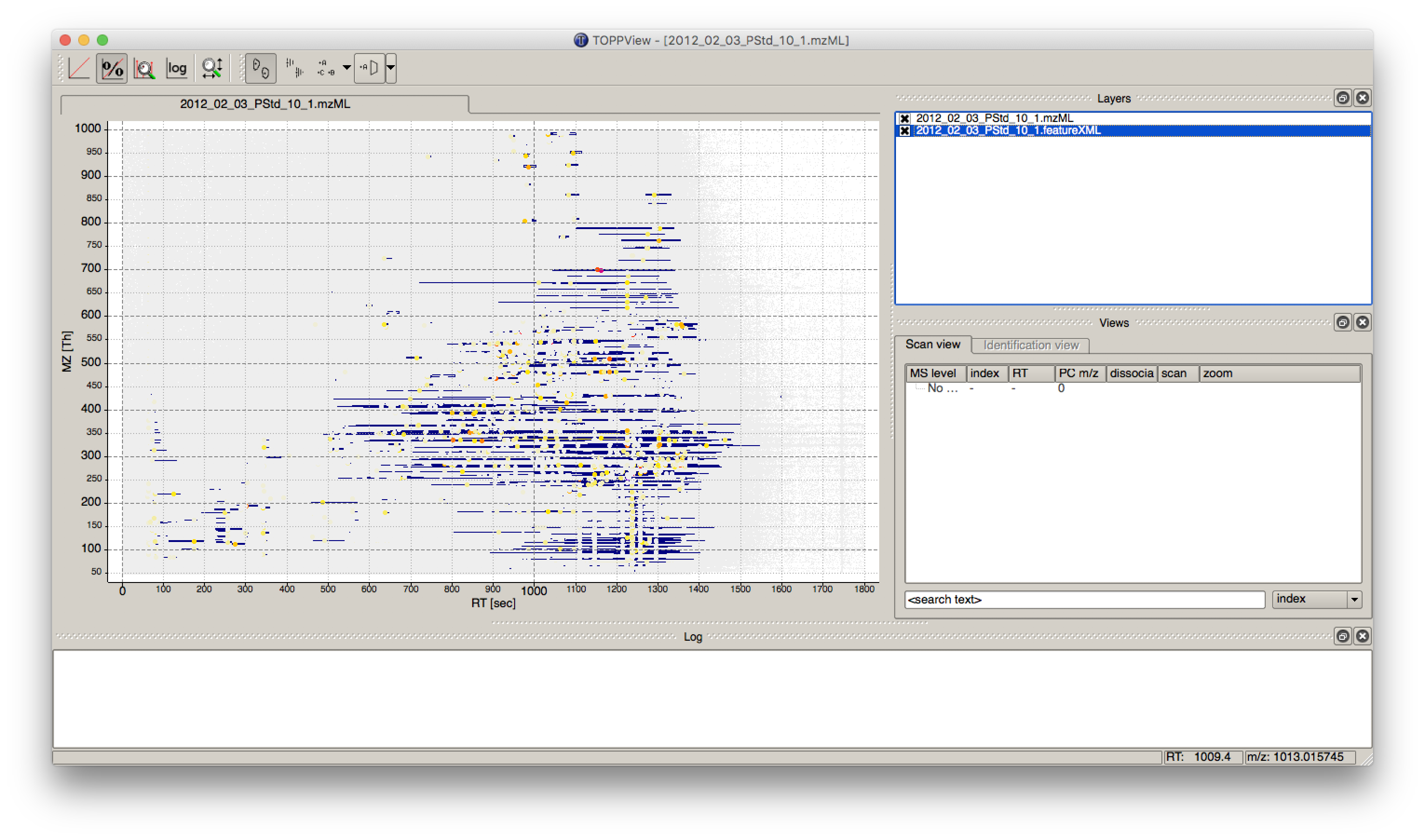
The output file .featureXML can be visualized with TOPPView on top of the used .mzML file - in a so-called layer - to look at the identified features.
First start TOPPView and open the example .mzML file (see Fig. 28). Afterwards open the .featureXML output as new layer (see Fig. 29). The overlay is depicted in Figure 30. The zoom of the .mzML - .featureXML overlay shows the individual mass traces and the assembly of those in a feature (see Fig. 31).
|
|---|
Figure 28: Opened .mzML in TOPPView |
|
|---|
Figure 29: Add new layer in TOPPView |
|
|---|
Figure 30: Overlay of the .mzML layer with the .featureXML layer |
|
|---|
Figure 31: Zoom of the overlay of the .mzML with the .featureXML layer. Here the individual isotope traces (blue lines) are assembled into a feature here shown as convex hull (rectangular box). |
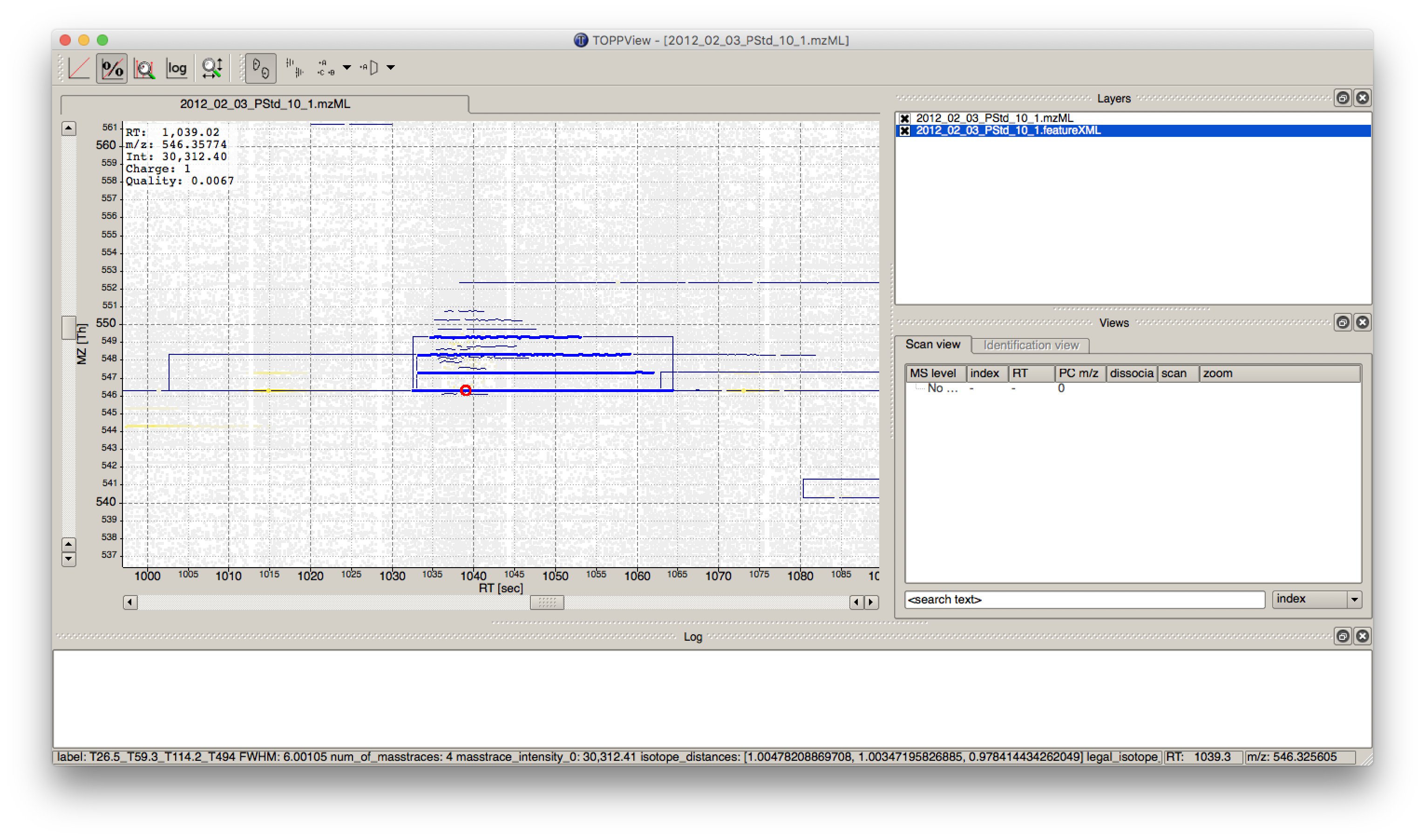
To correct precursor m/z values in centroided high-resolution data, one can add a HighResPrecursorMassCorrector node (Community Nodes > OpenMS > Mass Correction and Calibration) before the FeatureFinderMetabo node. The following parameter will be used to refine precursor masses by selecting the most intense centroided MS1 peak within a m/z tolerance of 10 ppm:
parameter |
value |
|---|---|
highest_intensity_peak → mz_tolerance → |
10.0 |
highest_intensity_peak → mz_tolerance_unit → |
ppm |
The parameters change the behavior of HighResPrecursorMassCorrector as follows:
mz_tolerance: The precursor mass tolerance to find the highest intensity MS1 peak.
mz_tolerance_unit: Unit of precursor mass tolerance.
The workflow can be extended for multi-file analysis by replacing the File Importer node with an Input Files node. Since HighResPrecursorMassCorrector and FeatureFinderMetabo operate on individual files, wrap them with a ZipLoopStart node before and a ZipLoopEnd node after.
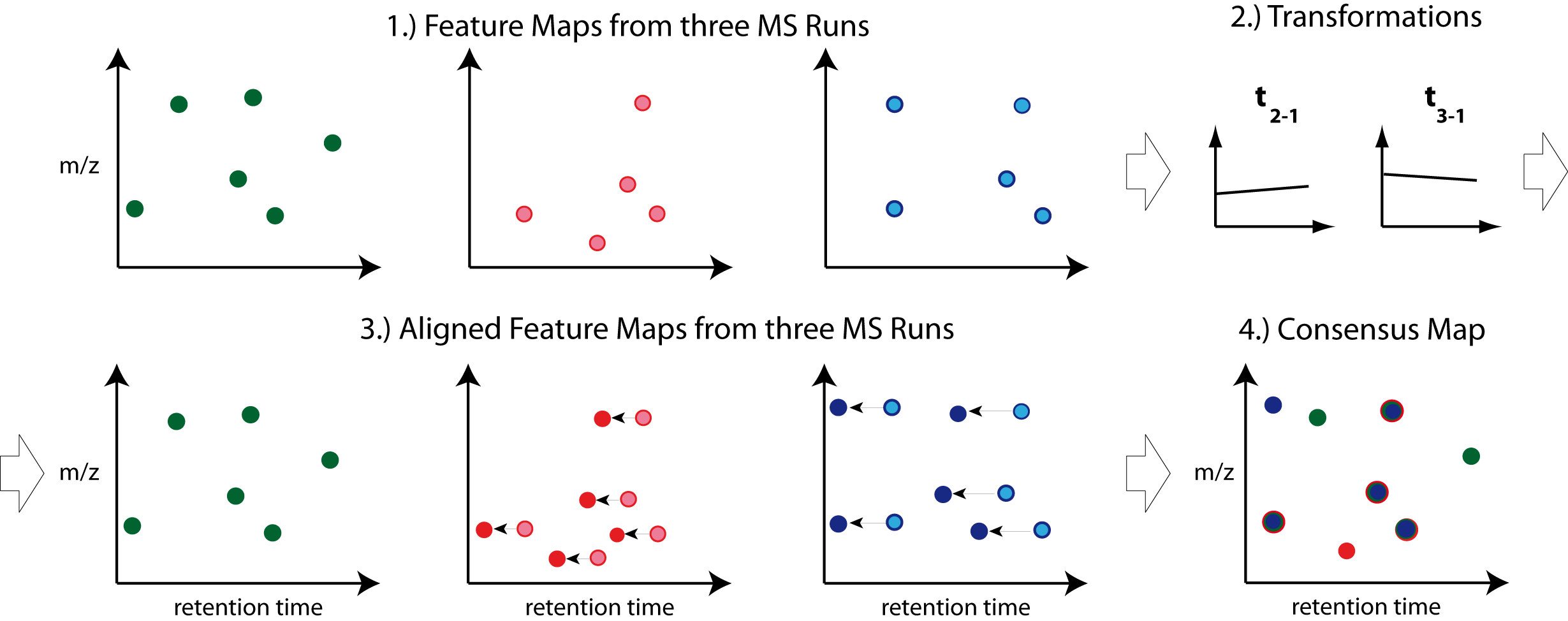
To facilitate the collection of features corresponding to the same compound ion across different samples, an alignment of the samples’ feature maps along retention time is often helpful. In addition to local, small-scale elution differences, one can often see constant retention time shifts across large sections between samples. We can use linear transformations to correct for these large-scale retention differences. This brings the majority of corresponding compound ions close to each other. Finding the correct corresponding ions is then faster and easier, as we don’t have to search as far around individual features.
|
|---|
Figure 32: The first feature map is used as a reference to which other maps are aligned. The calculated transformation brings corresponding features into close retention time proximity. Linking of these features form a so-called consensus features of a consensus map. |
After the
ZipLoopEndnode, add aMapAlignerPoseClusteringnode (Community Nodes>OpenMS>Map Alignment), set its Output Type to featureXML, and adjust the following settings:
parameter |
value |
|---|---|
algorithm → pairfinder → distance_MZ → max_difference |
10.0 |
algorithm → pairfinder → distance_MZ → unit |
ppm |
MapAlignerPoseClustering provides an algorithm to align the retention time scales of multiple input files, correcting shifts and distortions between them. Retention time adjustment may be necessary to correct for chromatography differences e.g. before data from multiple LC-MS runs can be combined (feature linking). The alignment algorithm implemented here is the pose clustering algorithm.
The parameters change the behavior of MapAlignerPoseClustering as follows:
max_difference: Features that have a larger m/z difference will never be paired.
unit: Unit used for the parameter distance_MZ max_difference, either Da or ppm.
The next step after retention time correction is the grouping of corresponding features in multiple samples. In contrast to the previous alignment, we assume no linear relations of features across samples. The used method is tolerant against local swaps in elution order.
|
|---|
Figure 33: Features A and B correspond to the same analyte. The linking of features between runs (indicated by an arrow) allows comparing feature intensities. |
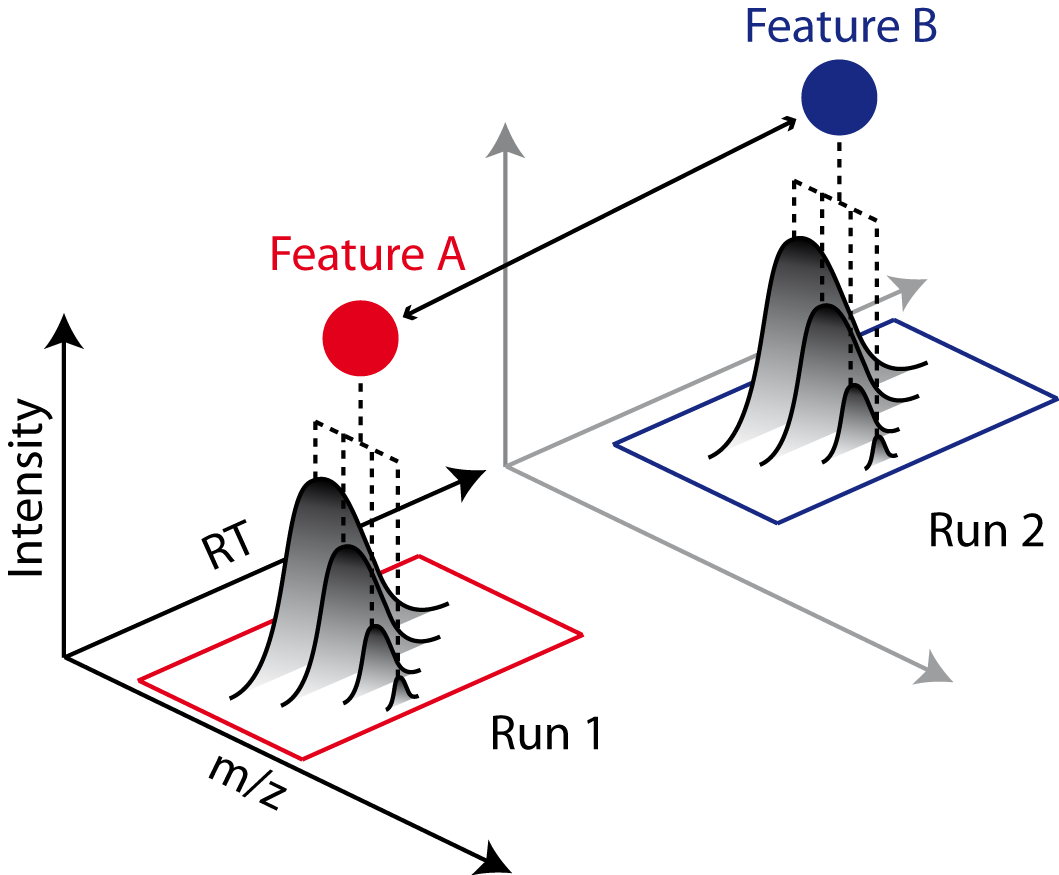
After the
MapAlignerPoseClusteringnode, add aFeatureLinkerUnlabeledKDnode (Community Nodes > OpenMS>Map Alignment) and adjust the following settings:|parameter|value| |:————|:——–| |algorithm → warp → enabled|false| |algorithm → link → rt_tol|30| |algorithm → link → mz_tol|10|
The parameters change the behavior of
FeatureLinkerUnlabeledKDas follows (similar to the parameters we adjusted forMapAlignerPoseClustering):warp → enabled: If set to true, feature RTs are warped using LOWESS transformation before linking (reported RTs in results will always be the original RTs).
link → rt_tol: Width of RT tolerance window (sec).
link → mz_tol: M/z tolerance.
After the
FeatureLinkerUnlabeledKDnode, add a TextExporter node (Community Nodes > OpenMS > File Handling).Add an
Output Foldernode and configure it with an output directory where you want to store the resulting files.Run the pipeline and inspect the output.
|
|---|
Figure 34: Label-free quantification workflow for metabolites. |

You should find a single, tab-separated file containing the information on where metabolites were found and with which intensities. You can also add Output Folder nodes at different stages of the workflow and inspect the intermediate results (e.g., identified metabolite features for each input map). The complete workflow can be seen in Figure 34. In the following section we will try to identify those metabolites.
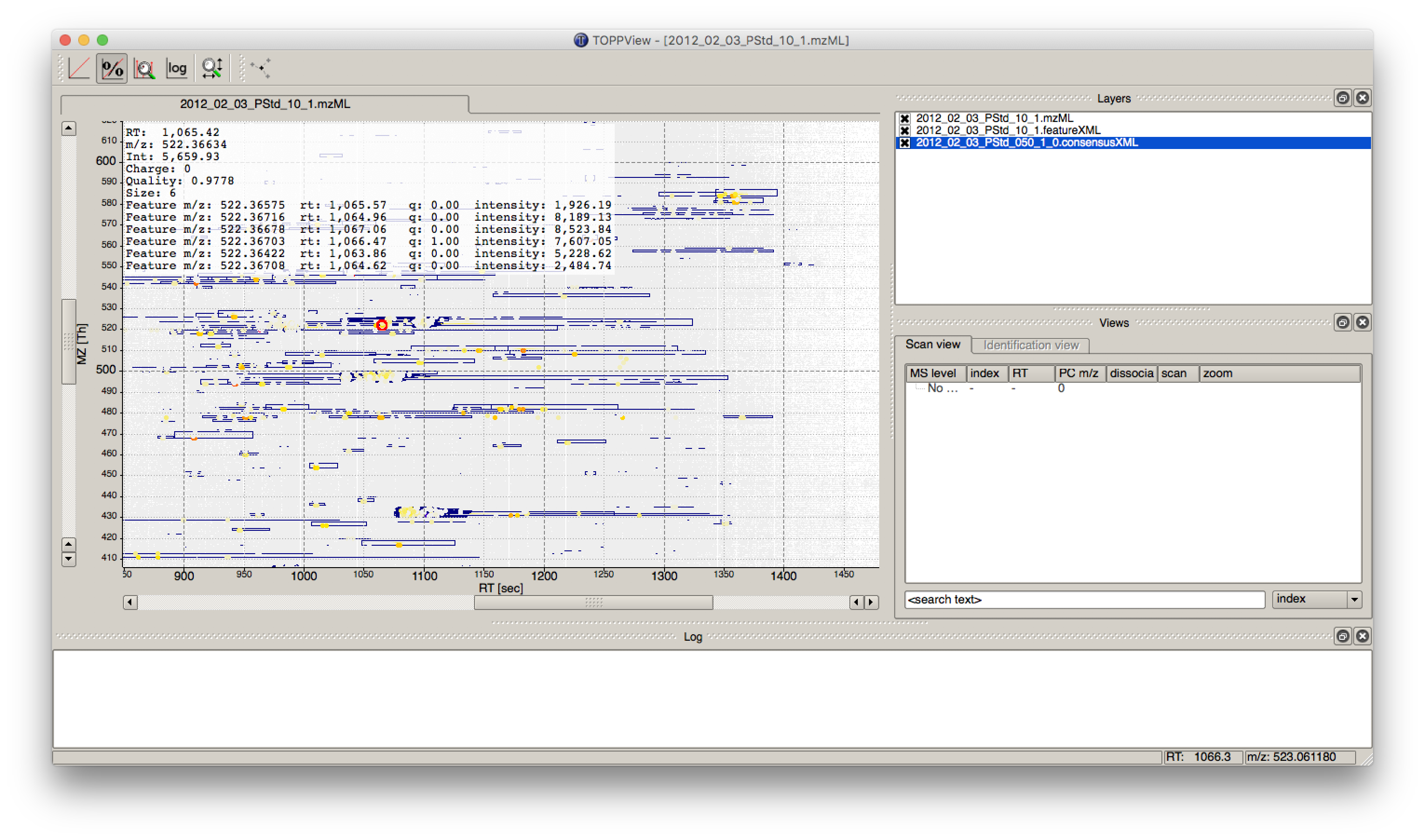
The FeatureLinkerUnlabeledKD output can be visualized in TOPPView on top of the input and output of the FeatureFinderMetabo (see Fig 35).
|
|---|
Figure 35: Visualization of .consensusXML output over the .mzML and .featureXML ’layer’. |
Basic metabolite identification¶
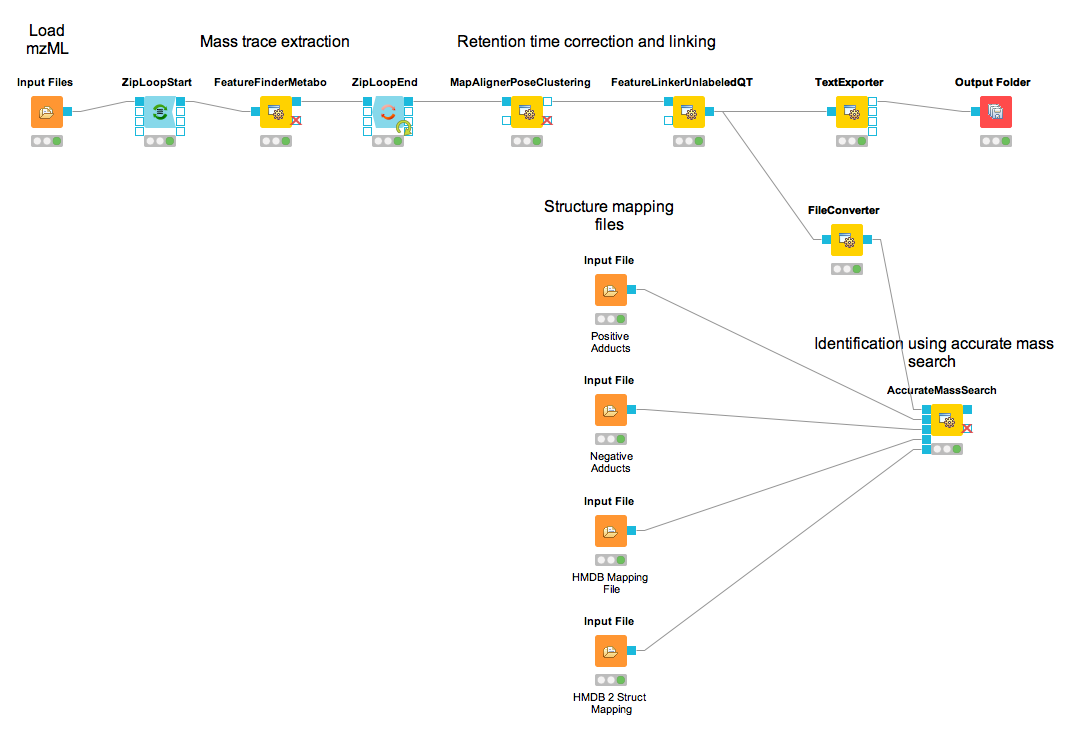
At the current state we found several metabolites in the individual maps but so far don’t know what they are. To identify metabolites, OpenMS provides multiple tools, including search by mass: the AccurateMassSearch node searches observed masses against the Human Metabolome Database (HMDB)[1], [2], [3]. We start with the workflow from the previous section (see Figure 34).
Add a FileConverter node (Community Nodes > OpenMS > File Handling) and connect the output of the FeatureLinkerUnlabeledKD to the incoming port.
Open the Configure dialog of the FileConverter node and select the tab OutputTypes. In the dropdown list for FileConverter.1.out select featureXML.
Add an AccurateMassSearch node (Community Nodes > OpenMS > Utilities) and connect the output of the FileConverter node to the first port of the AccurateMassSearch node.
Add four
File Importernodes and configure them with the following files:Example_Data▸Metabolomics▸databases▸PositiveAdducts.tsv This file specifies the list of adducts that are considered in the positive mode. Each line contains the formula and charge of an adduct separated by a semicolon (e.g. M+H;1+). The mass of the adduct is calculated automatically.Example_Data▸Metabolomics▸databases▸NegativeAdducts.tsv This file specifies the list of adducts that are considered in the negative mode analogous to the positive mode.Example_Data▸Metabolomics▸databases▸HMDBMappingFile.tsv This file contains information from a metabolite database in this case from HMDB. It has three (or more) tab-separated columns: mass, formula, and identifier(s). This allows for an efficient search by mass.Example_Data▸Metabolomics▸databases▸HMDB2StructMapping.tsv This file contains additional information about the identifiers in the mapping file. It has four tab-separated columns that contain the identifier, name, SMILES, and INCHI. These will be included in the result file. The identifiers in this file must match the identifiers in theHMDBMappingFile.tsv.
In the same order as they are given above connect them to the remaining input ports of the AccurateMassSearch node.
Add an
Output Foldernode and connect the first output port of the AccurateMassSearch node to theOutput Foldernode.
The result of the AccurateMassSearch node is in the mzTab format[4] so you can easily open it in a text editor or import it into Excel or KNIME, which we will do in the next section. The complete workflow from this section is shown in Figure 36.
|
|---|
Figure 36: Label-free quantification and identification workflow for metabolites. |
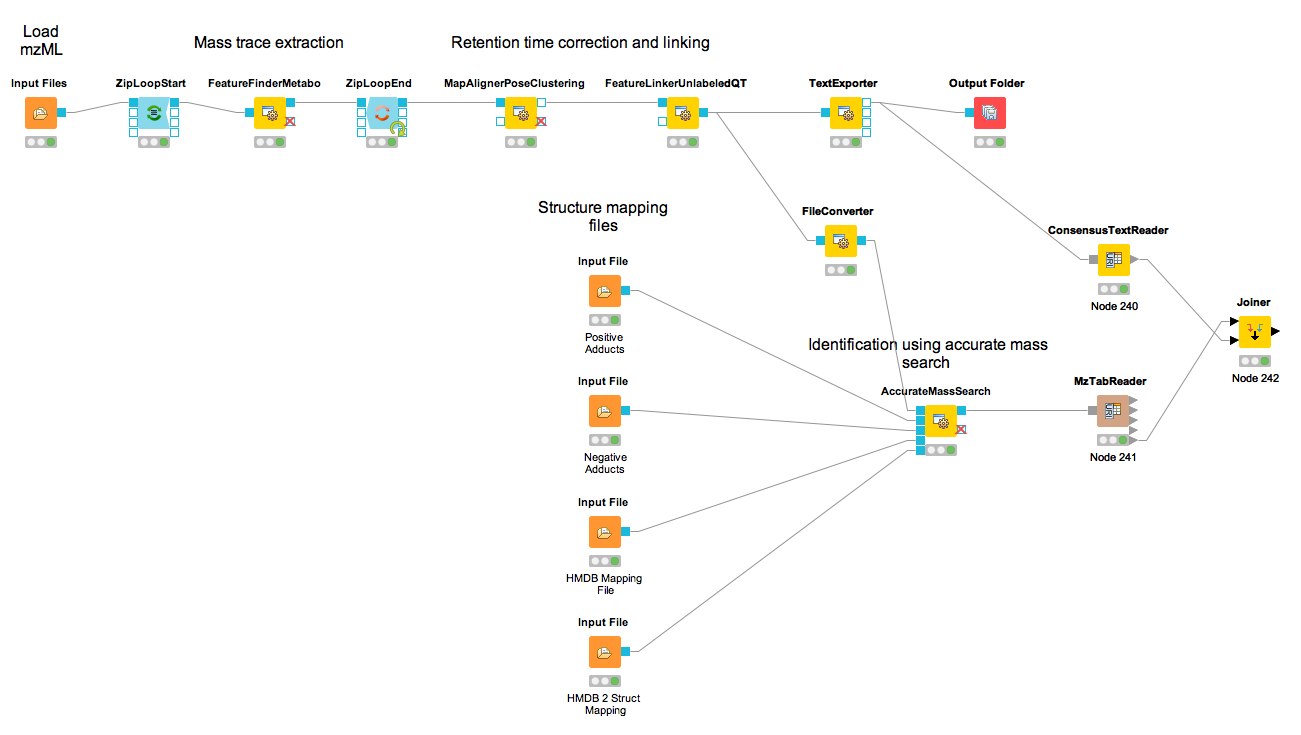
Convert your data into a KNIME table¶
The result from the TextExporter node as well as the result from the AccurateMassSearch node are files while standard KNIME nodes display and process only KNIME tables. To convert these files into KNIME tables we need two different nodes. For the AccurateMassSearch results, we use the MzTabReader node (Community Nodes > OpenMS > Conversion > mzTab) and its Small Molecule Section port. For the result of the TextExporter, we use the ConsensusTextReader (Community Nodes > OpenMS > Conversion).
When executed, both nodes will import the OpenMS files and provide access to the data as KNIME tables. The retention time values are exported as a list using the MzTabReader based on the current PSI-Standard. This has to be parsed using the SplitCollectionColumn, which outputs a ”Split Value 1” based on the first entry in the retention time list, which has to be renamed to retention time using the ColumnRename. You can now combine both tables using the Joiner node (Manipulation > Column > Split & Combine) and configure it to match the m/z and retention time values of the respective tables. The full workflow is shown in Figure 37.
|
|---|
Figure 37: Label-free quantification and identification workflow for metabolites that loads the results into KNIME and joins the tables. |
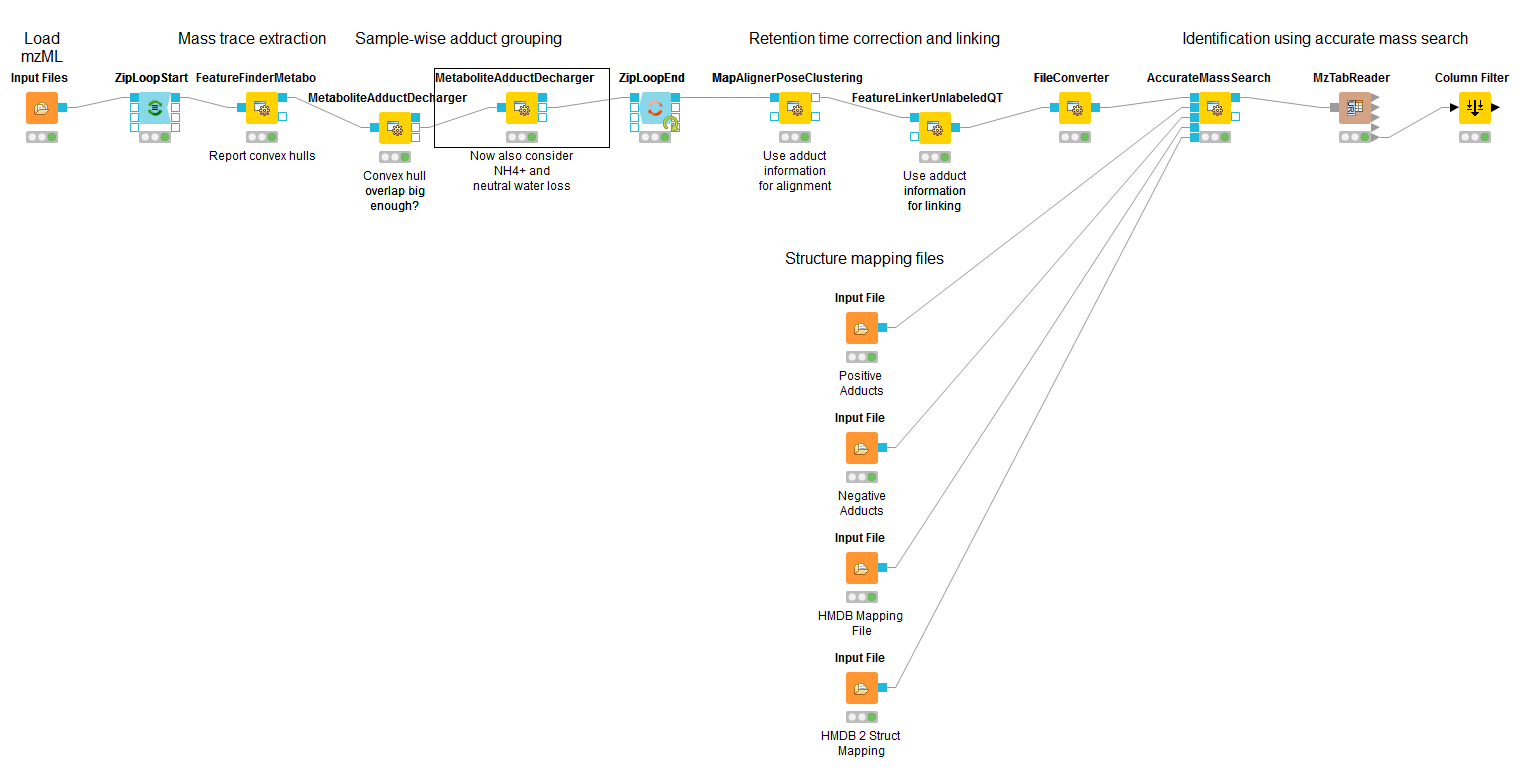
Adduct grouping¶
Metabolites commonly co-elute as ions with different adducts (e.g., glutathione+H, glutathione+Na) or with charge-neutral modifications (e.g., water loss). Grouping such related ions allows to leverage information across features. For example, a low-intensity, single-trace feature could still be assigned a charge and adduct due to a matching high-quality feature. Several OpenMS tools, such as AccurateMassSearch, can use this information to, for example, narrow down candidates for identification.
For this grouping task, we provide the MetaboliteAdductDecharger node. Its method explores the combinatorial space of all adduct combinations in a charge range for optimal explanations. Using defined adduct probabilities, it assigns co-eluting features having suitable mass shifts and charges those adduct combinations which maximize overall ion probabilities.
The tool works natively with featureXML data, allowing the use of reported convex hulls. On such a single-sample level, co-elution settings can be chosen more stringently, as ionization-based adducts should not influence the elution time: Instead, elution differences of related ions should be due to slightly differently estimated times for their feature centroids.
Alternatively, consensusXML data from feature linking can be converted for use, though with less chromatographic information. Here, the elution time averaging for features linked across samples, motivates wider co-elution tolerances.
The two main tool outputs are a consensusXML file with compound groups of related input ions, and a featureXML containing the input file but annotated with inferred adduct information and charges.
Options to respect or replace ion charges or adducts allow for example:
Heuristic but faster, iterative adduct grouping(MetaboliteAdductDecharger → MetaboliteFeatureDeconvolution → q_try set to “feature”) by chaining multiple MetaboliteAdductDecharger nodes with growing adduct sets, charge ranges or otherwise relaxed tolerances.
More specific feature linking (FeatureLinkerUnlabeledKD → algorithm → ignore_adduct set to “false”)
|
|---|
Figure 38: Metabolite Adduct Decharger adduct grouping workflow. |
Task
A modified metabolomics workflow with exemplary MetaboliteAdductDecharger use and parameters is provided in
Visualizing data¶
Now that you have your data in KNIME you should try to get a feeling for the capabilities of KNIME.
Task
Check out the Molecule Type Cast node (Chemistry > Translators) together with subsequent cheminformatics nodes (e.g. RDKit From Molecule(Community Nodes > RDKit > Converters)) to render the structural formula contained in the result table.
Task
Have a look at the Column Filter node to reduce the table to the interesting columns, e.g., only the Ids, chemical formula, and intensities.
Task
Try to compute and visualize the m/z and retention time error of the different feature elements (from the input maps) of each consensus feature. Hint: A nicely configured Math Formula (Multi Column) node should suffice.
Identifying Metabolites Using Spectral Libraries¶
Relying solely on accurate mass for metabolite identification can yield ambiguous results. In practice, additional information—such as retention time—is used to refine candidate selection. Beyond MS1-based features, tandem mass spectra (MS2) provide crucial structural insights. In this section, we explore how metabolites can be identified using a library of previously characterized spectra.
Annotating MS1 Features with Fragment Spectra¶
Before performing a spectral library search, we must annotate MS1-based features with their corresponding fragment spectra. To achieve this, we introduce the IDMapper node between MapAlignerPoseClustering and FeatureLinkerUnlabeledKD. Since IDMapper operates on a per-spectrum basis, we loop over the individual featureXML files using a ZipLoop. Additionally, as we aim to annotate MS spectra, we also connect the original mzML FileImporter node to the loop.
Originally designed for proteomics, IDMapper expects protein identifications. To bypass this requirement, we connect an empty ID file via a FileImporter node.
Filtering Unannotated MS1 Features¶
After linking features, we remove MS1-based features that lack fragment spectrum annotations. This is done by connecting a FileFilter node to the output of FeatureLinkerUnlabeledKD and enabling:
id → remove_unannotated_features = true
Generating Consensus Spectra¶
Next, we consolidate individual MS2 spectra into consensus spectra using the GNPSExport node, which outputs an mgf file containing the consensus spectra. To configure this step:
Connect the FileFilter node output to GNPSExport.
Also, connect the FileImporter node containing the original spectra.
Performing Spectral Library Matching¶
Now, we are ready to identify metabolites using the MetaboliteSpectralMatcher node:
Add a FileImporter node to load the spectral database.
Convert the
mgfoutput of GNPSExport intomzMLformat using the FileConverter node, ensuring:algorithm → merge_spectra = false
Add a MetaboliteSpectralMatcher node. Connect the FileConverter output (converted
mzML) to its query input, and the FileImporter (spectral database) to its library input.Read the resulting
mzTaboutput of MetaboliteSpectralMatcher into a KNIME table using the mzTabReader node.
This completes the workflow for spectral library-based metabolite identification.