About OpenMS¶
What is OpenMS¶
OpenMS is a free, open-source framework based on a C++ library with Python bindings. It is commonly used for liquid chromatography-mass spectrometry (LC-MS) data management and analyses. OpenMS provides an infrastructure for the rapid development of mass spectrometry related software as well as a rich toolset built on top of it. OpenMS is available under the three clause BSD licence and runs under Windows, macOS, and Linux operating systems.
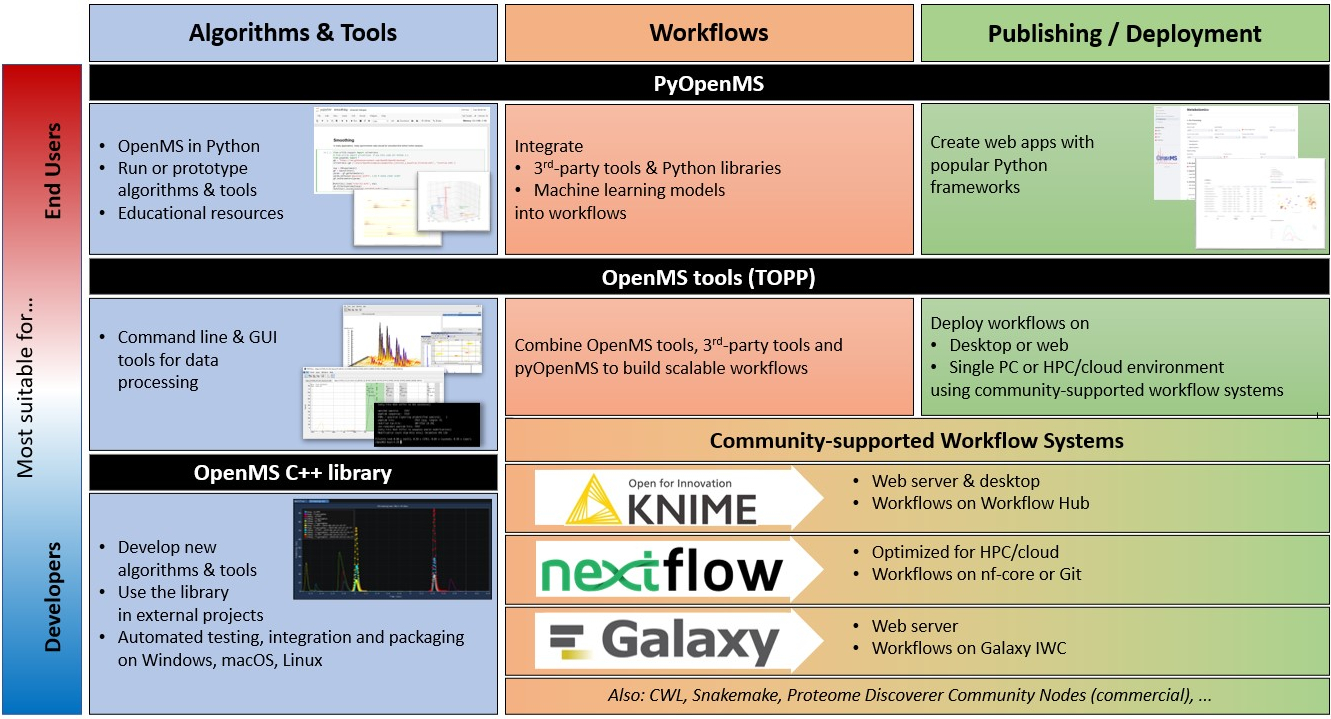
OpenMS developers can create new C++ algorithms and tools, while users can execute tools or implement new algorithms or scripts in Python. Workflows integrate pyOpenMS scripts and OpenMS tools with third-party tools and external Python libraries to create scalable data-processing pipelines. For deployment, users can use pyOpenMS with web frameworks or deploy workflows on desktop, high-performance computing (HPC) or cloud infrastructure using one of the community-supported workflow systems.
OpenMS supports the Proteomics Standard Initiative (PSI) formats for MS data. The main contributors of OpenMS are currently the Eberhard-Karls-Universität in Tübingen, the Freie Universität Berlin, and the University of Toronto.
Get involved¶
OpenMS is developed by a group of core developers and the community. You can help spreading the idea of open source mass spectrometry analysis by:
Contribute to the development by giving us your feedback about the OpenMS project on Discord or become active by developing new tools yourself.
Donate to the OpenMS project using our opencollective account. All donations will be used strictly to fund the development of Openms’s open source software, documentation, and community.
Promote OpenMS either online or in your work group.